Mutation Calling
The overview of the mutation calling pipeline that was used in Trisicell is provided below.
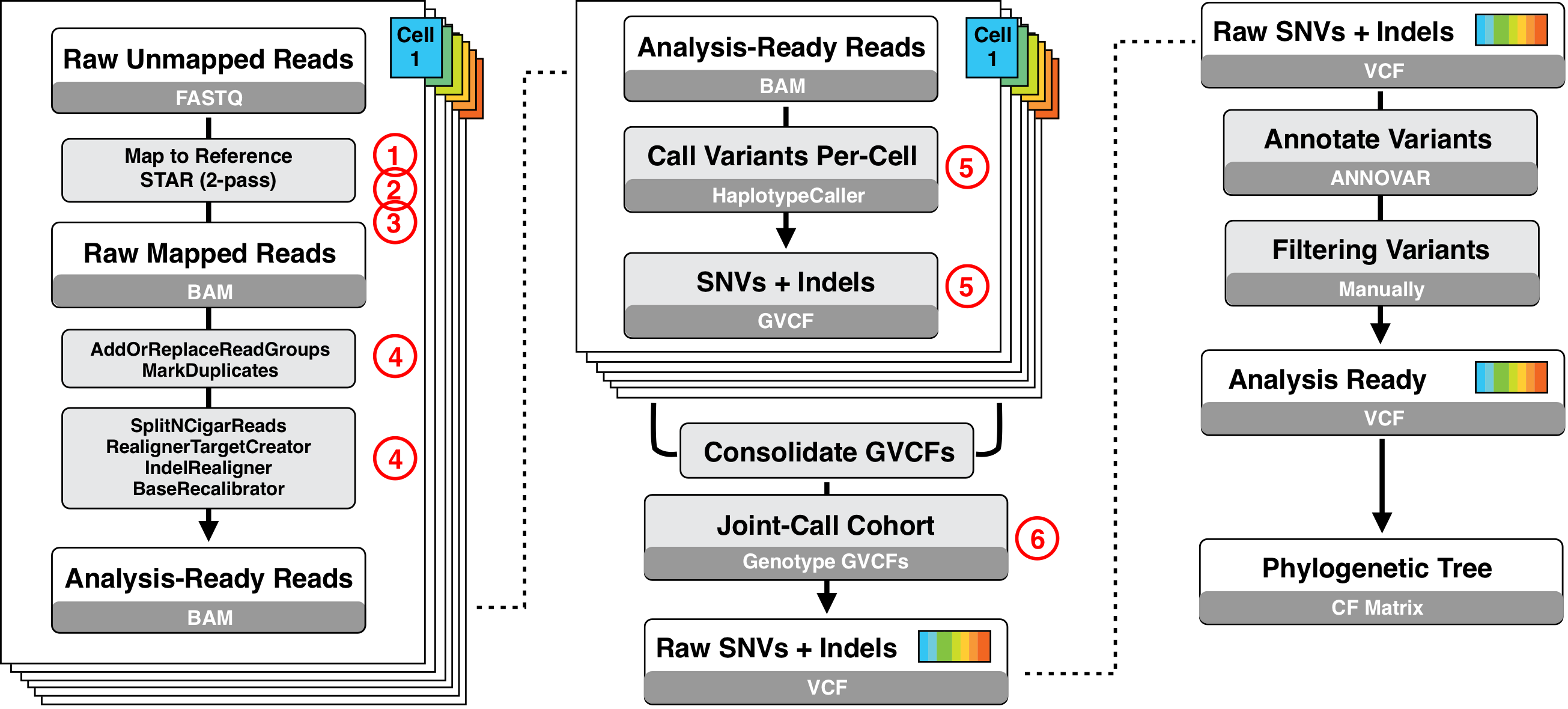
We provide a more detailed description of each of these steps.
Step 1:
STAR \
--runMode genomeGenerate \
--genomeDir {outdir}/{indexing_1} \
--genomeFastaFiles {ref} \
--sjdbGTFfile {gtf_file} \
--sjdbOverhang {readlength} \
--runThreadN {number_of_threads}
Step 2:
# for every `cell_id`
trim_galore \
--gzip \
--length 30 \
--fastqc \
--output_dir {outdir}/{cell_id} \
--paired \
{cell_id}/{fastq_file_1} {cell_id}/{fastq_file_2}
# for every `cell_id`
STAR \
--runMode alignReads \
--genomeDir {outdir}/{indexing1} \
--sjdbGTFfile {gtf_file} \
--outFilterMultimapNmax 1 \
--outSAMunmapped None \
--quantMode TranscriptomeSAM GeneCounts \
--runThreadN 1 \
--sjdbOverhang {read_length} \
--readFilesCommand zcat \
--outFileNamePrefix {outdir}/{cell_id}/ \
--readFilesIn {cell_id}/{fastq_file_1_trim_galore} {cell_id}/{fastq_file_2_trim_galore}
Step 3:
STAR \
--runMode genomeGenerate \
--genomeDir {outdir}/{indexing_2} \
--genomeFastaFiles {ref} \
--sjdbGTFfile {gtf_file} \
--sjdbFileChrStartEnd {all_cell_files [*SJ.out.tab]} \
--sjdbOverhang {read_length} \
--runThreadN {number_of_threads}
Step 4:
# for every `cell_id`
STAR \
--runMode alignReads \
--genomeDir {outdir}/{indexing_2} \
--readFilesCommand zcat \
--readFilesIn {cell_id}/{fastq_file_1_trim_galore} {cell_id}/{fastq_file_2_trim_galore} \
--outFileNamePrefix {outdir}/{cell_id}/ \
--limitBAMsortRAM 30000000000 \
--outSAMtype BAM SortedByCoordinate \
--sjdbGTFfile {gtf_file} \
--outFilterMultimapNmax 1 \
--outSAMunmapped None \
--quantMode TranscriptomeSAM GeneCounts \
--runThreadN 1 \
--sjdbOverhang {read_length}
# for every `cell_id`
java -Xmx90g -jar PICARD.jar \
AddOrReplaceReadGroups \
INPUT={outdir}/{cell_id}/Aligned.sortedByCoord.out.bam \
OUTPUT={outdir}/{cell_id}/dedupped.bam \
SORT_ORDER=coordinate \
RGID={cell_id} \
RGLB=trancriptome \
RGPL=ILLUMINA \
RGPU=machine \
RGSM={cell_id}
# for every `cell_id`
java -Xmx90g -jar PICARD.jar \
MarkDuplicates \
INPUT={outdir}/{cell_id}/dedupped.bam \
OUTPUT={outdir}/{cell_id}/rg_added_sorted.bam \
METRICS_FILE={outdir}/{cell_id}/output.metrics \
VALIDATION_STRINGENCY=SILENT \
CREATE_INDEX=true
# for every `cell_id`
java -Xmx90g -jar GATK.jar \
-T SplitNCigarReads \
-R {ref} \
-I {outdir}/{cell_id}/rg_added_sorted.bam \
-o {outdir}/{cell_id}/split.bam \
-rf ReassignOneMappingQuality \
-RMQF 255 \
-RMQT 60 \
-U ALLOW_N_CIGAR_READS
# for every `cell_id`
java -Xmx90g -jar GATK.jar \
-T RealignerTargetCreator \
-R {ref} \
-I {outdir}/{cell_id}/split.bam \
-o {outdir}/{cell_id}/indel.intervals \
-known {db_snps_indels} \
-U ALLOW_SEQ_DICT_INCOMPATIBILITY
# for every `cell_id`
java -Xmx90g -jar GATK.jar \
-T IndelRealigner \
-R {ref} \
-I {outdir}/{cell_id}/split.bam \
-o {outdir}/{cell_id}/realign.bam \
-targetIntervals {outdir}/{cell_id}/indel.intervals \
-known {db_snps_indels}
# for every `cell_id`
java -Xmx90g -jar GATK.jar \
-T BaseRecalibrator \
-R {ref} \
-I {outdir}/{cell_id}/realign.bam \
-o {outdir}/{cell_id}/recal.table \
-knownSites {db_snps_indels}
# for every `cell_id`
java -Xmx90g -jar GATK.jar \
-T PrintReads \
-R {ref} \
-I {outdir}/{cell_id}/realign.bam \
-o {outdir}/{cell_id}/output.bam \
-BQSR {outdir}/{cell_id}/recal.table
Step 5:
# for every `cell_id`
java -Xmx90g -jar GATK.jar \
-T HaplotypeCaller \
-R {ref} \
-I {outdir}/{cell_id}/output.bam \
-o {outdir}/{cell_id}/HaplotypeCaller.g.vcf \
-dontUseSoftClippedBases \
-stand_call_conf 20 \
--dbsnp {db_snps_indels} \
-ERC GVCF
Step 6:
java -Xmx90g -jar GATK.jar \
-T GenotypeGVCFs \
-R {ref} \
-V {all_cell_files [*HaplotypeCaller.g.vcf]} \
-o {outdir}/jointcalls.g.vcf \
-nt {number_of_threads} \
--disable_auto_index_creation_and_locking_when_reading_rods